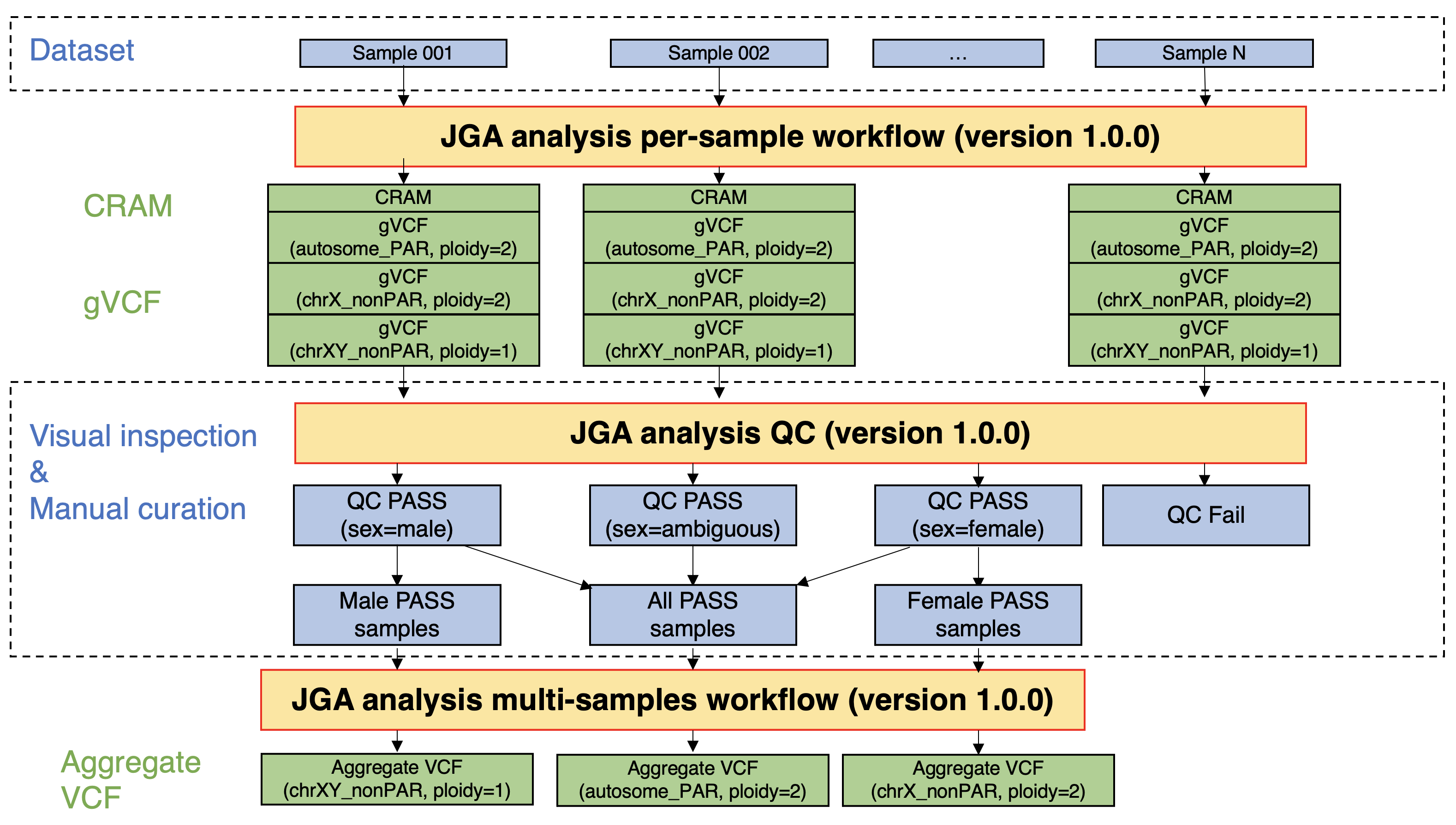
生殖系列(germline)の全ゲノムシークエンスデータの加工
JGAでは、データの汎用性を考慮し、次世代シークエンサー(next-generation sequencer:NGS)から出力されたデータはFASTQもしくはBAM形式で登録いただいている。そのためデータ利用者は、FASTQ/BAM形式のデータをダウンロードし、データ利用者ごとにバリアント検出等のデータ加工を行う必要がある。
JGAに登録されているデータの利便性向上のため、JGAに登録されている生殖系列の全ゲノムシークエンス(whole genome sequencing:WGS)データについて共通ワークフローを用いて加工し、アライメント結果(CRAM)、サンプルごとのバリアント検出結果(gVCF)、データセットごとのバリアント検出結果(aggregate VCF)を計算した。計算結果である加工データは元のデータに紐づけた形でJGAに登録されており、元データの利用をNBDCヒトデータ審査委員会より承認されたデータ利用者が希望する場合、加工データも利用可能となる。
データ加工方法
生殖系列のWGSデータの加工は、国立遺伝学研究所スーパーコンピュータシステムの個人ゲノム解析区画の計算資源と、国立研究開発法人科学技術振興機構 NBDC 事業推進部が開発したJGAデータ加工ワークフロー(jga-analysis)を用いて行った。JGAデータ加工ワークフローはGATK best practice - Germline short variant discovery(SNPs + Indels)に則り、ワークフロー言語であるCommon Workflow Language(CWL)を用いて実装されている。JGAデータ加工ワークフローのソースコードは GitHub 上で公開している(https://github.com/ddbj/jga-analysis)。
データ加工の具体的な手順は以下の通り。
-
JGA analysis per-sample workflow(version 1.0.0)の実行
- このワークフローは、FASTQファイルを入力として、reference genome(GRCh38) にアライメントし、サンプルごとにバリアント検出を行う。アライメント結果(CRAM)、サンプルごとのバリアント検出結果(gVCF)、後のステップで使用する品質管理指標(CRAM-level and gVCF-level metrics)が出力される。
- このワークフローは、次のステップから構成される。
- Alignment(FASTQ to SAM):bwa mem(version 0.7.15)
- SAM to BAM:GATK SortSam(version 4.1.0.0)
- MarkDuplicates:GATK MarkDuplicates(version 4.1.0.0)
- BQSR(optional):GATK BaseRecalibrator(version 4.1.0.0)、GATK ApplyBQSR(version 4.1.0.0)
- BAM to CRAM:samtools view -C(version 1.9)、samtools index(version 1.9)
- Calculate cram-level metrics:samtools idxstats(version 1.9)、samtools flagstat(version 1.9)、GATK CollectBaseDistributionByCycle(version 4.2.0.0)、GATK CollectWgsMetrics(version 4.2.0.0)
- Variant call:GATK HaplotypeCaller -ERC GVCF(version 4.1.0.0)、bgzip(version 1.9)、tabix(version 1.9)
- Calculate gVCF-level metrics:bcftools stats(version 1.9)
- バリアント検出時のパラメータである ploidy は次のように設定している。
- 常染色体および pseudoautosomal region(PAR) 領域:ploidy=2
- X染色体のnon-PAR領域:ploidy=2(女性) および ploidy=1(男性)
- Y染色体の non-PAR 領域:ploidy=1(男性)
- このワークフロー実行時には、サンプルの性別に関わらず、「常染色体および PAR 領域:ploidy=2」、「X染色体のnon-PAR領域:ploidy=2」、「X染色体のnon-PAR領域:ploidy=1」、「Y染色体の non-PAR 領域:ploidy=1」のすべてのパターンでバリアント検出を行い、gVCF ファイルを出力しました。品質管理ステップにおいてサンプルの性別をゲノムデータから推定し、どの ploidy で計算した gVCF を使用するかを条件分岐するようにしました。
-
JGA analysis QC(version 1.0.0)の実行。
- このプログラムは、JGA analysis per-sample workflow で計算した結果得られる cram-level & gVCF-level metrics を図示化して、品質管理(quality control:QC)を行う。QC の具体的な項目は以下の通り。
- 常染色体および PAR 領域の mean coverage の分布をヒストグラムにより確認し、mean coverage が外れ値を示すサンプルを除外
- 「(X 染色体の non-PAR 領域の mean coverage)/ (常染色体および PAR 領域の mean coverage)」と「(Y 染色体の non-PAR 領域の mean coverage)/ (常染色体および PAR 領域の mean coverage)」を指標として、サンプルの性別を推定
- このプログラムは、JGA analysis per-sample workflow で計算した結果得られる cram-level & gVCF-level metrics を図示化して、品質管理(quality control:QC)を行う。QC の具体的な項目は以下の通り。
-
JGA analysis multi-samples workflow(version 1.0.0)の実行
- このワークフローは、複数サンプルの gVCF ファイルを入力として、joint call と variant quality score recalibration (VQSR)を行い、データセットごとのバリアント検出結果(aggregate VCF)を出力する。その後、aggregate VCF から個人ごとのデータを削除した統計情報(sites-only aggregate VCF)を計算し、出力する。
- このワークフローは、次のステップから構成される。
- Joint call:GATK GenomicsDBImport(version 4.2.0.0)、GATK GenotypeGVCFs(version 4.2.0.0)、GATK VariantFiltration(version 4.2.0.0)
- VQSR:GATK GatherVcfs(version 4.2.0.0)、GATK VariantRecalibrator --mode INDEL(version 4.2.0.0)、GATK VariantRecalibrator --mode SNP(version 4.2.0.0)、GATK ApplyVQSR -model INDEL(version 4.2.0.0)、GATK ApplyVQSR -model SNP(version 4.2.0.0)、bgzip(version 1.9)、bcftools index -t(version 1.9)
- Calculate aggregate VCF metrics:GATK CollectVariantCallingMetrics(version 4.2.0.0)
- Calculate sites-only aggregate VCF:GATK MakeSitesOnlyVcf(version 4.2.0.0)、bgzip(version 1.9)、bcftools index -t(version 1.9)
- このワークフロー実行時には、JGA analysis QC ステップで推定されたサンプルの性別に基づいて、ワークフローに入力する gVCF ファイルを選択するよう設定している。
- 常染色体および PAR 領域:QC ステップを通過したすべてのサンプルを対象に、「常染色体および PAR 領域:ploidy=2」の条件で計算されたgVCFファイルをワークフローの入力とした。
- X染色体のnon-PAR領域:QC ステップを通過したすべてのサンプルのうち、性別が女性と推定されたサンプルを対象に、「X染色体のnon-PAR領域:ploidy=2」の条件で計算されたgVCFファイルをワークフローの入力とした。また、QC ステップを通過したすべてのサンプルのうち、性別が男性と推定されたサンプルを対象に、「X染色体のnon-PAR領域:ploidy=1」の条件で計算された gVCF ファイルをワークフローの入力とした。
- Y染色体の non-PAR 領域:QC ステップを通過したすべてのサンプルのうち、性別が男性と推定されたサンプルを対象に、「Y染色体の non-PAR 領域:ploidy=1」の条件で計算された gVCF ファイルをワークフローの入力とした。
図:生殖系列の全ゲノムシークエンスデータ加工のフロー