Processing of whole genome sequencing analysis data (germline)
In JGA, most of the whole genome sequencing (WGS) data are registered in the FASTQ/BAM format, because of the versatility of the data. Accordingly, the data users have to download the WGS data, followed by data processing by themselves.
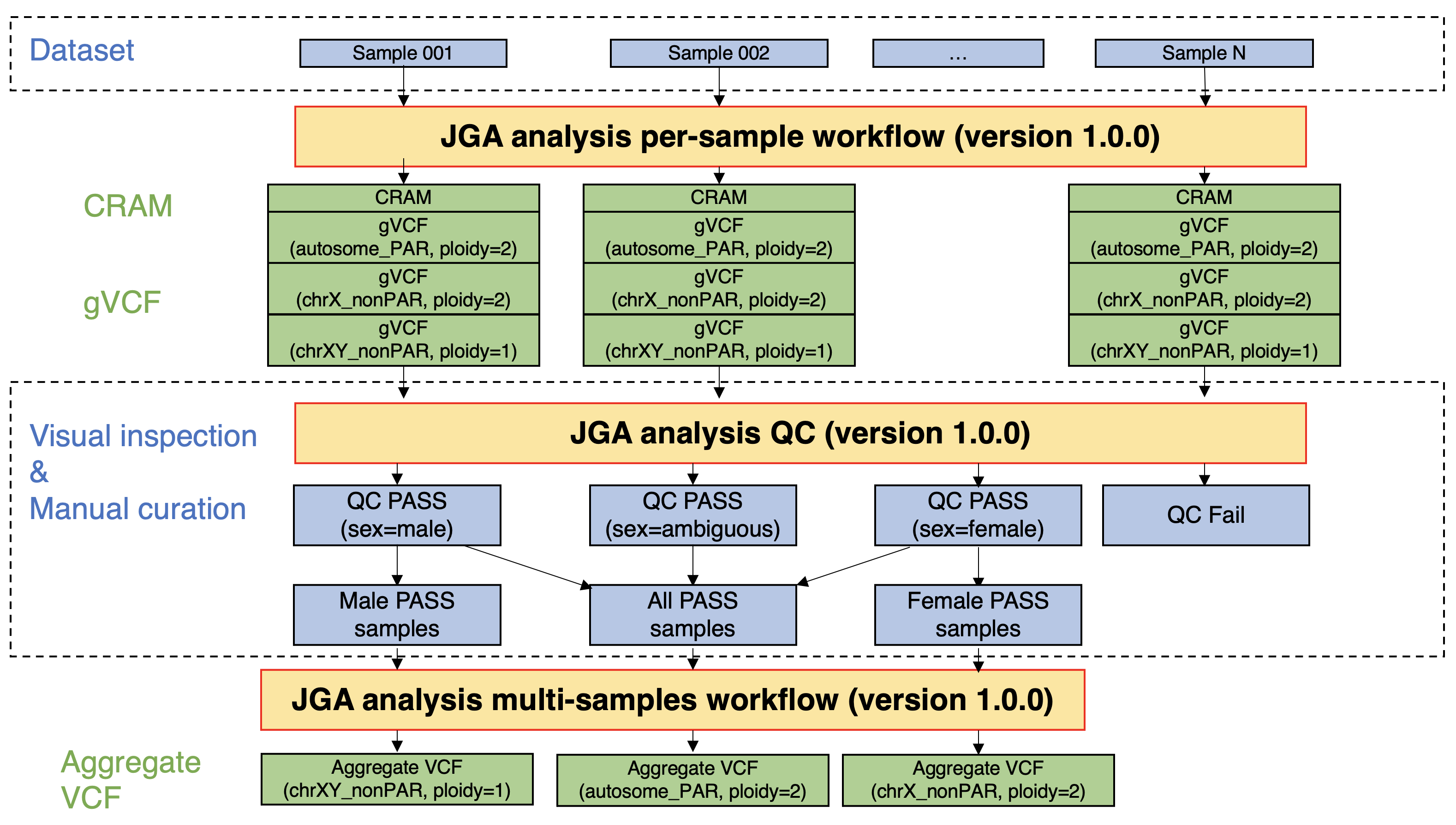
To improve the convenience of the data users, germline WGS data registered in JGA were processed in a certain workflow, and alignment results (CRAM), variant call results per sample (gVCF), and variant call results per dataset (aggregated VCF) were calculated. The post-processing data have been registered in the JGA linked to the original data, and the data users who were approved by the NBDC Data Access Committee can also download the post-processing data.
Methods of data processing
The germline WGS data were processed using the computational resources of the Personal Genome Analysis Section of the National Institute of Genetics Supercomputer System, and the JGA data analysis workflow developed by the Department of NBDC Program, Japan Science and Technology Agency. The analysis workflow was implemented based on the GATK best practice - Germline short variant discovery (SNPs + Indels) using the Common Workflow Language (CWL). The source code of the workflow is available on GitHub at https://github.com/ddbj/jga-analysis.
-
Perform JGA analysis per-sample workflow (version 1.0.0)
- This workflow takes FASTQ files as input, aligns them to the reference genome (GRCh38), and performs variant call per sample. The alignment results (CRAM), variant call results per sample (gVCF), and quality control metrics (CRAM-level and gVCF-level metrics) used in later steps are output.
- This workflow consists of the following steps:
- Alignment (FASTQ to SAM): bwa mem (version 0.7.15)
- SAM to BAM: GATK SortSam (version 4.1.0.0)
- MarkDuplicates: GATK MarkDuplicates (version 4.1.0.0)
- BQSR (optional): GATK BaseRecalibrator (version 4.1.0.0), GATK ApplyBQSR (version 4.1.0.0)
- BAM to CRAM: samtools view -C (version 1.9), samtools index (version 1.9)
- Calculate cram-level metrics: samtools idxstats (version 1.9), samtools flagstat (version 1.9), GATK CollectBaseDistributionByCycle (version 4.2.0.0), GATK CollectWgsMetrics (version 4.2.0.0)
- Variant call: GATK HaplotypeCaller -ERC GVCF (version 4.1.0.0), bgzip (version 1.9), tabix (version 1.9)
- Calculate gVCF-level metrics: bcftools stats (version 1.9)
- The ploidy for variant call was set as follows:
- Autosomes and pseudoautosomal regions (PARs): ploidy=2
- Non-PARs on the X chromosome: ploidy=2 (female) and ploidy=1 (male)
- Non-PARs on the Y chromosome: ploidy=1 (male)
- All the four patterns ("Autosomes and PARs: ploidy=2", "Non-PARs on the X chromosome: ploidy=2", "Non-PARs on the X chromosome: ploidy=1", and "Non-PARs on the Y chromosome: ploidy=1") were executed regardless of the sex of the sample. The sex of the sample is estimated from the genomic data in the quality control (QC) step.
-
Perform JGA analysis QC (version 1.0.0).
- This program performs QC by visualizing the CRAM- and gVCF-level metrics calculated by the abovementioned JGA analysis per-sample workflow. QC items were listed below:
- The distribution of mean coverage for autosomes and PARs were visualized by histograms, and samples with outliers in mean coverage were excluded.
- The sex of the sample was estimated using "(mean coverage of non-PARs on the X chromosome) / (mean coverage of autosomes and PARs)" and "(mean coverage of non-PARs on the Y chromosome) / (mean coverage of autosomes and PARs)".
- This program performs QC by visualizing the CRAM- and gVCF-level metrics calculated by the abovementioned JGA analysis per-sample workflow. QC items were listed below:
-
Perform JGA analysis multi-samples workflow (version 1.0.0).
- This workflow takes multiple gVCF files as input, performs joint call and variant quality score recalibration (VQSR), and outputs variant call results per dataset (aggregate VCF). The summarized data (sites-only aggregate VCF) was then calculated.
- This workflow consists of the following steps:
- Joint call: GATK GenomicsDBImport (version 4.2.0.0), GATK GenotypeGVCFs (version 4.2.0.0), GATK VariantFiltration (version 4.2.0.0)
- VQSR: GATK GatherVcfs (version 4.2.0.0), GATK VariantRecalibrator --mode INDEL (version 4.2.0.0), GATK VariantRecalibrator --mode SNP (version 4.2.0.0), GATK ApplyVQSR -model INDEL (version 4.2.0.0), GATK ApplyVQSR -model SNP (version 4.2.0.0), bgzip (version 1.9), bcftools index -t (version 1.9)
- Calculate aggregate VCF metrics: GATK CollectVariantCallingMetrics (version 4.2.0.0)
- Calculate sites-only aggregate VCF: GATK MakeSitesOnlyVcf (version 4.2.0.0), bgzip (version 1.9), bcftools index -t (version 1.9)
- When this workflow was executed, the gVCF files to be input into the workflow were selected based on the sex of the samples as estimated in the JGA analysis QC step.
- Autosomes and PARs: For all samples that passed the QC step, gVCF files calculated under the condition "Autosomes and PARs: ploidy=2" were used as input for the workflow.
- Non-PARs of the X chromosome: For all female samples that passed the QC step, gVCF files calculated under the condition "Non-PARs of the X chromosome: ploidy=2" were used as input for the workflow. For all male samples that passed the QC step, gVCF files calculated under the condition "Non-PARs of the X chromosome: ploidy=1" were used as input for the workflow.
- Non-PARs of the Y chromosome: For all male samples that passed the QC step, gVCF files calculated under the condition "Non-PARs of the Y chromosome: ploidy=1" were used as input for the workflow.
Figure: A flow of germline WGS data processing