

We are currently receiving a large number of Applications for data submission, and the review process is taking longer than usual.
We sincerely apologize for the delay and kindly ask for your understanding.
When submitting an application, we would greatly appreciate it if you could allow sufficient time for the processing.
About NBDC Human Database
An enormous amount of human data is being generated with advances in next-generation sequencing and other analytical technologies. We therefore need rules and mechanisms for organizing and storing such data and for effectively utilizing them to make progress in the life sciences.
To promote sharing and utilization of human data while considering the protection of personal information, the Database Center for Life Science (DBCLS) of the Joint Support-Center for Data Science Research, Research Organization of Information and Systems (ROIS-DS) created a platform for sharing various data generated from human specimens, which are available for publicly access in cooperation with the DNA Data Bank of Japan![]() .
.
You can apply to use or submit human data through this website.
Violators of the guidelines who have not submitted a report on the deletion of Controlled-access data shall be disclosed here.
What's New
Contact
For inquiries regarding the guidelines and the submission or use of data in the NBDC Human Database, please use this "Contact Form".
Contact
The National Bioscience Database Center (NBDC), the Japan Science and Technology Agency (JST)
NBDC Data Sharing Subcommittee Office
NBDC Human Data Review Board Office
5-3, Yonbancho, Chiyoda-ku, Tokyo 102-0081, Japan
Tel. +81-3-5214-8491
Fax. +81-3-5214-8470
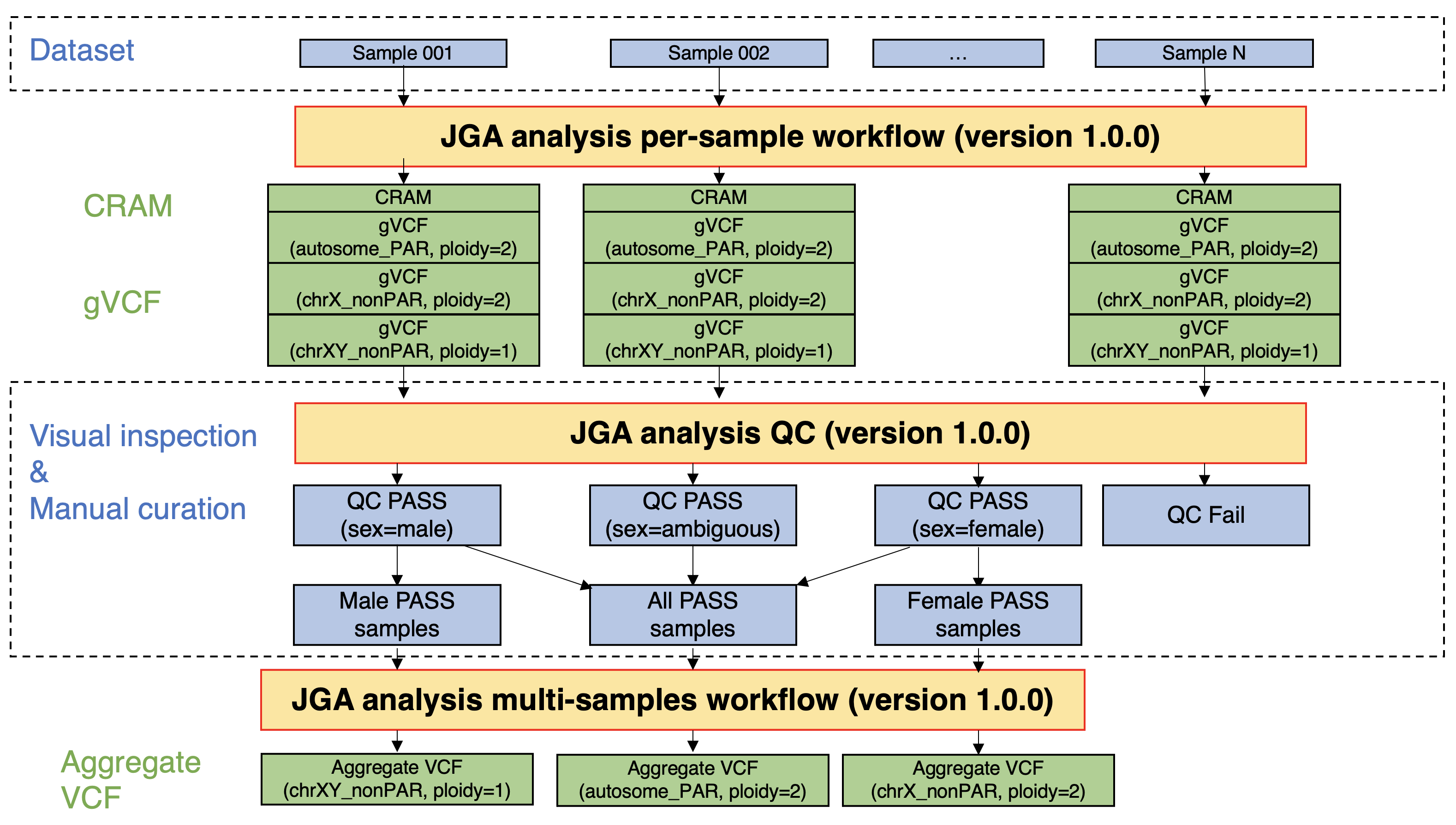
Processing of whole genome sequencing analysis data (germline)
In JGA, most of the whole genome sequencing (WGS) data are registered in the FASTQ/BAM format, because of the versatility of the data. Accordingly, the data users have to download the WGS data, followed by data processing by themselves.
To improve the convenience of the data users, germline WGS data registered in JGA were processed in a certain workflow, and alignment results (CRAM), variant call results per sample (gVCF), and variant call results per dataset (aggregated VCF) were calculated. The post-processing data have been registered in the JGA linked to the original data, and the data users who were approved by the NBDC Data Access Committee can also download the post-processing data.
Methods of data processing
The germline WGS data were processed using the computational resources of the Personal Genome Analysis Section of the National Institute of Genetics Supercomputer System, and the JGA data analysis workflow developed by the Department of NBDC Program, Japan Science and Technology Agency. The analysis workflow was implemented based on the GATK best practice - Germline short variant discovery (SNPs + Indels) using the Common Workflow Language (CWL). The source code of the workflow is available on GitHub at https://github.com/ddbj/jga-analysis.
-
Perform JGA analysis per-sample workflow (version 1.0.0)
- This workflow takes FASTQ files as input, aligns them to the reference genome (GRCh38), and performs variant call per sample. The alignment results (CRAM), variant call results per sample (gVCF), and quality control metrics (CRAM-level and gVCF-level metrics) used in later steps are output.
- This workflow consists of the following steps:
- Alignment (FASTQ to SAM): bwa mem (version 0.7.15)
- SAM to BAM: GATK SortSam (version 4.1.0.0)
- MarkDuplicates: GATK MarkDuplicates (version 4.1.0.0)
- BQSR (optional): GATK BaseRecalibrator (version 4.1.0.0), GATK ApplyBQSR (version 4.1.0.0)
- BAM to CRAM: samtools view -C (version 1.9), samtools index (version 1.9)
- Calculate cram-level metrics: samtools idxstats (version 1.9), samtools flagstat (version 1.9), GATK CollectBaseDistributionByCycle (version 4.2.0.0), GATK CollectWgsMetrics (version 4.2.0.0)
- Variant call: GATK HaplotypeCaller -ERC GVCF (version 4.1.0.0), bgzip (version 1.9), tabix (version 1.9)
- Calculate gVCF-level metrics: bcftools stats (version 1.9)
- The ploidy for variant call was set as follows:
- Autosomes and pseudoautosomal regions (PARs): ploidy=2
- Non-PARs on the X chromosome: ploidy=2 (female) and ploidy=1 (male)
- Non-PARs on the Y chromosome: ploidy=1 (male)
- All the four patterns ("Autosomes and PARs: ploidy=2", "Non-PARs on the X chromosome: ploidy=2", "Non-PARs on the X chromosome: ploidy=1", and "Non-PARs on the Y chromosome: ploidy=1") were executed regardless of the sex of the sample. The sex of the sample is estimated from the genomic data in the quality control (QC) step.
-
Perform JGA analysis QC (version 1.0.0).
- This program performs QC by visualizing the CRAM- and gVCF-level metrics calculated by the abovementioned JGA analysis per-sample workflow. QC items were listed below:
- The distribution of mean coverage for autosomes and PARs were visualized by histograms, and samples with outliers in mean coverage were excluded.
- The sex of the sample was estimated using "(mean coverage of non-PARs on the X chromosome) / (mean coverage of autosomes and PARs)" and "(mean coverage of non-PARs on the Y chromosome) / (mean coverage of autosomes and PARs)".
- This program performs QC by visualizing the CRAM- and gVCF-level metrics calculated by the abovementioned JGA analysis per-sample workflow. QC items were listed below:
-
Perform JGA analysis multi-samples workflow (version 1.0.0).
- This workflow takes multiple gVCF files as input, performs joint call and variant quality score recalibration (VQSR), and outputs variant call results per dataset (aggregate VCF). The summarized data (sites-only aggregate VCF) was then calculated.
- This workflow consists of the following steps:
- Joint call: GATK GenomicsDBImport (version 4.2.0.0), GATK GenotypeGVCFs (version 4.2.0.0), GATK VariantFiltration (version 4.2.0.0)
- VQSR: GATK GatherVcfs (version 4.2.0.0), GATK VariantRecalibrator --mode INDEL (version 4.2.0.0), GATK VariantRecalibrator --mode SNP (version 4.2.0.0), GATK ApplyVQSR -model INDEL (version 4.2.0.0), GATK ApplyVQSR -model SNP (version 4.2.0.0), bgzip (version 1.9), bcftools index -t (version 1.9)
- Calculate aggregate VCF metrics: GATK CollectVariantCallingMetrics (version 4.2.0.0)
- Calculate sites-only aggregate VCF: GATK MakeSitesOnlyVcf (version 4.2.0.0), bgzip (version 1.9), bcftools index -t (version 1.9)
- When this workflow was executed, the gVCF files to be input into the workflow were selected based on the sex of the samples as estimated in the JGA analysis QC step.
- Autosomes and PARs: For all samples that passed the QC step, gVCF files calculated under the condition "Autosomes and PARs: ploidy=2" were used as input for the workflow.
- Non-PARs of the X chromosome: For all female samples that passed the QC step, gVCF files calculated under the condition "Non-PARs of the X chromosome: ploidy=2" were used as input for the workflow. For all male samples that passed the QC step, gVCF files calculated under the condition "Non-PARs of the X chromosome: ploidy=1" were used as input for the workflow.
- Non-PARs of the Y chromosome: For all male samples that passed the QC step, gVCF files calculated under the condition "Non-PARs of the Y chromosome: ploidy=1" were used as input for the workflow.
Figure: A flow of germline WGS data processing
About Data Processing
For the convenience of data users, alignment data, variant call data, and statistical data, which are processed by a certain workflow on data deposited to the NBDC Human Database as controlled-access data (original data), can be used together with the original data if the user who is permitted to use the original data by the Human Data Review Board and wishes to use the processed data as well. The processed data are placed in a way that they are connected to the original data, and are indicated as "Processed by JGA" in the title of the Analysis and Dataset. When publishing analysis results including processed data, please include the accession number of the original data in the article.
How to process the data:
- Whole genome sequencing analysis data (germline)
- Data were processed by using of GATK Best Practice - Germline short variant discovery (SNPs and Indels)
- Workflow Source Code (jga-analysis): https://github.com/ddbj/jga-analysis
- List of processed data
- Imputation reference panel
- Reference panels were created by using of bcftools (version 1.9) and beagle-bref3 (version 28Jun21.220)
- Workflow Source Code (imputation-server-wf):
- bcftools-index-t.cwl: https://github.com/ddbj/imputation-server-wf/blob/main/Tools/bcftools-index-t.cwl
- beagle-bref3.cwl: https://github.com/ddbj/imputation-server-wf/blob/main/Tools/beagle-bref3.cwl
- List of processed data
* In such data processing conducted by the DBCLS and the Bioinformation and DDBJ Center, the data will not be used for any purposes other than those of activities to promote the use of the NBDC human database.
Target datasets
| JGA dataset | Study title | #. Samples | Date of data processing | Remarks |
|---|---|---|---|---|
| JGAD000252 | Cancer genomics for elucidation of molecular mechanisms of carcinogenecis and progression in lung cancer (hum0068) | 21 | Per-sample:2021-12-27 QC:2022-01-06 Joint-call:2022-01-26 |
This dataset includes the whole genome sequencing analysis (WGS) data of tumor and matched control samples. WGS data of the matched control (germline) were processed. Dataset ID of the processed data: JGAD000670 |
| JGAD000235 | Genome sequencing analysis for colorectal cancer (hum0159) | 10 | Per-sample:2021-12-27 QC:2022-01-06 Joint-call:2022-01-31 |
This dataset includes WGS data of tumor and matched control samples. WGS data of the matched control (germline) were processed. Dataset ID of the processed data: JGAD000689 |
| JGAD000234 | Genome sequencing analysis for hepatoblastoma (hum0161) | 33 | Per-sample:2021-12-27 QC:2022-01-06 Joint-call:2022-01-31 |
This dataset includes WGS data of tumor and matched control samples. WGS data of the matched control (germline) were processed. Dataset ID of the processed data: JGAD000688 |
| JGAD000335 | Collection and transfer of human tumor samples and research using genomic information Transfer of existing samples and research using genomic information Reseach of searching gene mutations in gastrointestinal chronic inflammatory diseases (hum0201) |
14 | Per-sample:2021-12-27 QC:2022-01-06 Joint-call:2022-01-06 |
This dataset includes WGS data of tumor and matched control samples. WGS data of the matched control (germline) were processed. Dataset ID of the processed data: JGAD000687 |
| JGAD000220 | Bio Bank Japan project (hum0014) | 1,026 | Per-sample:2022-01-16 QC:2022-02-24 |
Data processing was performed on the WGS data of 1,026 individuals registered to the Biobank Japan Project from FY 2003 to FY 2007. Dataset ID of the processed data: JGAD000690 |
| JGAD000220 | Bio Bank Japan project (hum0014) | 1,026 | Joint-call: 2023-01-30 (autosome and chrX PAR regions; BOTHSEX), 2023-02-25 (chrX non-PAR regions; FEMALE samples), 2023-03-02 (chrXY non-PAR regions; MALE samples) |
Data processing was performed on the WGS data of 1,026 individuals registered to the Biobank Japan Project from FY 2003 to FY 2007. Joint-call was performed for genotype calls. Dataset ID of the processed data: JGAD000758 |
*In order to use the processed data, it is necessary to apply the application for data use of the original data (Dataset IDs are shown on the most left side of the above table).
![]()